PHYS 5120 - 计算能源材料和电子结构模拟 Lecture
I:
这些内容共同构成了量子化学中用于计算分子电子结构的基石——Hartree-Fock (HF) 自洽场 (SCF) 方法。
核心概念:Hartree-Fock (HF) 近似
- 目标:求解多电子体系(如分子)的薛定谔方程。但这个方程非常复杂,无法精确求解。
- 核心思想 (HF 近似):
- 波恩-奥本海默近似:假定原子核固定不动,我们只关心电子的运动。
- 单电子近似:这是 HF 近似最关键的一步。它假设每个电子的运动只受到一个“平均场”的影响,这个平均场是由原子核和其他所有电子共同产生的。
- 反对称性:电子是费米子,必须满足泡利不相容原理。HF 方法通过一个称为“斯莱特行列式 (Slater Determinant)”的数学工具来构造总波函数,以自动满足这个要求。
⬅基本算符与方程
1. Hartree-Fock
方程:f̂ |φi> = εi |φi>
它在形式上是一个本征方程。
|φi>(分子轨道):这是我们要求解的第i个单电子波函数(或称为轨道)。f̂(Fock 算符):这是一个等效的单电子哈密顿算符。它代表了一个电子在原子核和所有其他电子的“平均场”中所感受到的总能量。εi(轨道能量):这是第i个分子轨道的能量,即电子占据该轨道时的能量。
2. Fock
算符的构成:f̂ = ĥ₁(r) + Σ[j] (2Ĵj(r) - K̂j(r))
这个公式详细定义了 Fock 算符 f̂ 是由什么组成的。
ĥ₁(r)(核心哈密顿算符):这是“单电子”部分,与电子间的相互作用无关。它只包括:- 电子的动能:白板上的
-(ħ²/2m)∇²项。 - 电子与所有原子核的吸引势能:白板上的
-Σ[I] (Z_I e² / |r - R_I|)项。
- 电子的动能:白板上的
Σ[j] (2Ĵj(r) - K̂j(r))(双电子相互作用):这是描述电子i与其他所有电子j之间平均相互作用的项(求和Σ[j]遍历所有被占据的轨道)。Ĵj(r)(库仑算符):- 物理意义:描述了电子
i与轨道j上的电子云(密度为|φj|²)之间的经典静电排斥。 - 白板上的定义:
Ĵj(ψ) = <φj | e² / |r - r'| | φj> ψ。这意味着Ĵj作用在一个函数ψ(r)上时,它会计算ψ(r)与φj(r')之间的平均排斥能。
- 物理意义:描述了电子
K̂j(r)(交换算符):- 物理意义:这是一个纯粹的量子力学效应,没有经典对应。它源于波函数的反对称性要求(泡利不相容原理)。它修正了电子“自我排斥”的错误(因为
Ĵj包含了j=i的情况),并降低了自旋平行电子相遇的概率,从而降低了能量。 - 白板上的定义:
K̂j(ψ) = <φj | e² / |r - r'| | ψ> φj。注意ψ和φj在积分符号内的位置发生了“交换”,因此得名。
- 物理意义:这是一个纯粹的量子力学效应,没有经典对应。它源于波函数的反对称性要求(泡利不相容原理)。它修正了电子“自我排斥”的错误(因为
- 系数 “2”:在闭壳层(Closed-Shell)HF
方法中,我们假设每个分子轨道
j都被两个自旋相反的电子(一个自旋向上α,一个自旋向下β)占据。因此,Ĵj的排斥作用要乘以 2。而K̂j的交换作用只发生在自旋相同的电子之间,因此只有一个K̂j被减去(例如,自旋向上的电子i只与自旋向上的电子j发生交换)。
3. HOMO / LUMO
- HOMO (Highest Occupied Molecular
Orbital):最高占据分子轨道。这是 HF 计算得到的
εi能量中,能量最高但仍被电子占据的那个轨道。 - LUMO (Lowest Unoccupied Molecular
Orbital):最低未占分子轨道。这是
εi能量中,能量最低但没有电子占据的那个轨道。 - 意义:HOMO 和 LUMO 统称为“前线轨道”。它们的能量差(HOMO-LUMO Gap)和形状在化学反应中至关重要,决定了分子倾向于从哪里给出电子 (HOMO) 和从哪里接受电子 (LUMO)。
矩阵化 (Roothaan-Hall 方法)
直接求解上面的 Hartree-Fock 方程(它是微分-积分方程)非常困难。C. C. J. Roothaan 和 G. G. Hall 提出了一种将其转换为标准矩阵代数问题的方法。
1. LCAO 展开
我们假设未知的分子轨道 |φi>
可以由一组已知的原子轨道 (Atomic Orbitals, AOs)
|χμ> 线性组合而成:
|φi> = Σ[μ] Cμi |χμ> 其中 Cμi
是我们要解的系数。
2. Roothaan-Hall
方程:F C = S C ε
这是将 LCAO 展开代入 HF 方程后得到的矩阵方程。
F(Fock 矩阵):Fμν = <χμ | f̂ | χν>。这是 Fock 算符f̂在原子轨道基组下的矩阵表示。C(系数矩阵):矩阵的每一列i都是一个分子轨道φi的展开系数Cμi。S(重叠矩阵):Sμν = <χμ | χν>。它描述了原子轨道基函数之间的重叠程度。如果基函数是“正交”的,S就是单位矩阵。ε(轨道能量矩阵):这是一个对角矩阵,对角线上的元素εi就是我们要求的分子轨道能量。
3. Fock
矩阵元的计算:Fμν = Hμν^core + Gμν
这是求解的核心,它把 F 矩阵的计算分为两部分:
- ①
Hμν^core = <χμ | ĥ₁ | χν>(核心哈密顿矩阵元):- 物理意义:这是“单电子”部分,只包含电子动能和电子-原子核吸引能。
- 计算:这部分在整个计算过程中只用计算一次,因为它不依赖于电子的分布。
Gμν(双电子积分项):- 物理意义:这是“双电子”排斥部分,对应于
Σ[j] (2Ĵj - K̂j)。 - 问题:计算
Gμν需要知道库仑和交换算符,而这些算符又依赖于分子轨道|φj>,而|φj>又是由我们要求解的系数C决定的。这就形成了一个“鸡生蛋,蛋生鸡”的循环问题。
- 物理意义:这是“双电子”排斥部分,对应于
4. 密度矩阵与 SCF 循环
为了解决这个循环问题,我们引入了密度矩阵
P。
- ③ 密度矩阵 (Density Matrix)
P:- 白板上的定义:
Pαβ = 2 Σ[j=1 to N/2] Cαj* Cβj - 物理意义:
P描述了电子在原子轨道基组上的分布情况。它由系数矩阵C构造。
- 白板上的定义:
- 使用
P构建 Fock 矩阵F:- 白板上的公式:
Fμν = Hμν^core + Σ[αβ] Pαβ * (...)(白板上(...)部分代表了(μν|αβ) - 1/2(μα|νβ)这样的双电子积分项)。 - 关键点:
F矩阵(代表电子间的相互作用)现在被表示为密度矩阵P(代表电子的分布)的函数。F = F(P)。
- 白板上的公式:
总结:自洽场 (SCF) 的完整流程
完整地描述了 “自洽场” (Self-Consistent Field, SCF) 的计算流程:
- 第 0 步:选择原子轨道基组
|χμ>,并计算所有不变的积分,如重叠矩阵S和核心哈密顿矩阵H^core。 - 第 1 步 (猜测):对系数矩阵
C做一个初始猜测(例如,通过一个更简单的方法得到),并用它来构建一个初始的密度矩阵P^(0)。 - 第 2 步 (构建):使用当前的密度矩阵
P^(k)来构建 Fock 矩阵F^(k)。Fμν = Hμν^core + Gμν(P^(k)) - 第 3 步 (求解):求解 Roothaan-Hall 矩阵方程
F^(k) C^(k+1) = S C^(k+1) ε^(k+1),得到一组新的系数矩阵C^(k+1)和新的轨道能量ε^(k+1)。 - 第 4 步 (更新):使用新的系数
C^(k+1)来计算一个新的密度矩阵P^(k+1)。 - 第 5 步
(检查自洽):比较新的密度矩阵
P^(k+1)和旧的密度矩阵P^(k)。- 如果
P^(k+1)和P^(k)几乎相同(即“自洽”了),说明我们找到的电子分布P所产生的“平均场”F,反过来再求解这个F得到的电子分布恰好就是P本身。计算收敛,循环结束。 - 如果它们不相同,就令
k = k+1,返回第 2 步,用新的P继续迭代。
- 如果
这个迭代过程,就是 Hartree-Fock 自洽场方法的核心。
II:
两个关键部分:
- 1.:继续详细推导如何使用密度矩阵 (Density Matrix) 来构建 Fock 矩阵中的双电子相互作用项。
- 2.:展示了如何从数学上求解 Roothaan-Hall 方程
(
F C = S C ε),这是一个“广义本征值问题”,并将其转换为计算机可以轻松处理的“标准本征值问题”。
Fock 矩阵元的最终形式
这部分是整个 HF-SCF
方法中最核心的数学推导之一。它展示了如何将复杂的双电子相互作用(Gμν)表示为密度矩阵
P 和一堆预先计算好的积分的乘积。
Pαβ = 2 Σ[j=1 to N/2] Cαj* Cβj- 重申密度矩阵
P的定义。它由系数矩阵C构建。
- 重申密度矩阵
- 推导
③:<χμ | Σ[j] ... | χν>- 这一长串推导(从
③开始,一直到= Σ[αβ] Pαβ (...))是白板上最复杂的部分。 - 目标:计算 Fock 矩阵的双电子部分
Gμν = <χμ | Σ[j] (2Ĵj - K̂j) | χν>。 - 步骤:
- 将分子轨道
|φj>用原子轨道|χ>展开:|φj> = Σ[α] Cαj |χα>。 - 将这个展开式代入
Ĵj和K̂j的积分定义中。 - 这会产生涉及四个原子轨道基函数的积分,称为双电子积分
(Two-Electron Integrals),通常写作
(μν|αβ)。 - 经过复杂的代数重排(将
C和Σ[j]重新组合),推导发现Gμν可以被写成:Gμν = Σ[αβ] Pαβ * [ (μν|αβ) - 1/2 (μα|νβ) ](μν|αβ)是库仑积分。(μα|νβ)是交换积分。
- 将分子轨道
- 这一长串推导(从
Fμν = Hμν^core + Σ[αβ] Pαβ (...)- 最终的 Fock 矩阵元公式。
Fμν(Fock 矩阵)=Hμν^core(核心哈密顿矩阵,只算一次)+Gμν(双电子排斥项)。- 关键在于
Gμν是密度矩阵P的线性函数。 - 这完美地建立了 SCF 的循环关系:
C→P→F→ 求解F得到新的C。
Roothaan-Hall 方程的求解问题
这部分转向了一个纯粹的数学(线性代数)问题:如何求解我们建立的矩阵方程。
S† F C = C ε(笔误)- 白板上划掉的
S† F C = C ε及其旁边的推导(S†F)† = ...看起来像是一个错误的尝试或旁注,试图探索这个矩阵的厄米性 (Hermiticity)。 - 正确的方程(在它下面)是
F C = S C ε。
- 白板上划掉的
F C = S C ε(Roothaan-Hall 方程)- 问题:这不是一个“标准本征值问题”
(
A x = λ x),因为在等式右边多了一个重叠矩阵S。S不是单位矩阵I,因为原子轨道基组|χ>通常不是正交的。 - 术语:这被称为“广义本征值问题”。
- 问题:这不是一个“标准本征值问题”
(
S is positive definite(S 是正定矩阵)- 这是一个关键的数学性质。
S是正定的,意味着它所有的本征值(λi)都大于零。 - 意义:因为
S是正定的,所以它保证是可逆的(S⁻¹存在),并且我们可以对它进行“开方”,即找到S^(1/2)和S^(-1/2)。
- 这是一个关键的数学性质。
S = R† R或S = U† Λ U(S 的分解)- 这是对
S矩阵进行对角化或分解的标准方法。 S = U† Λ U是谱分解:U是S的本征向量矩阵。Λ是由S的本征值λi组成的对角矩阵。
- 这是对
S^(1/2) = U† √Λ U和S^(-1/2) = U† (1/√Λ) U- (右侧白板上有
S^(-1/2)的定义)。 - 这是利用
S的分解来定义它的1/2次方和-1/2次方矩阵。√Λ就是简单地将对角矩阵Λ上的每个元素λi都开方。
- (右侧白板上有
变换为标准本征值问题
这部分展示了如何利用 S^(-1/2) 矩阵来“清除”方程中的
S,将其变为标准本征值问题。这个过程称为正交化
(Orthogonalization)。
- 目标:将
F C = S C ε变换为F' C' = C' ε的形式。 - 定义新的系数矩阵
C': 我们定义一组新的、在正交化基组下的系数C',它与原始系数C的关系是:C' = S^(1/2) C(因此C = S^(-1/2) C') - 代入原方程: 将
C = S^(-1/2) C'代入F C = S C ε:F (S^(-1/2) C') = S (S^(-1/2) C') ε - 两边左乘
S^(-1/2):(S^(-1/2) F S^(-1/2)) C' = (S^(-1/2) S S^(-1/2)) C' ε - 简化:
- 右侧:如白板所示,
S^(-1/2) S S^(-1/2) = S^(-1/2) S^(1/2) S^(1/2) S^(-1/2) = I(单位矩阵)。 - 左侧:我们定义一个新的、变换后的 Fock 矩阵
F':F' = S^(-1/2) F S^(-1/2)
- 右侧:如白板所示,
- 最终的标准本征值方程:
F' C' = C' ε
总结:SCF 循环的完整计算步骤
一个完整的 SCF 迭代步骤如下:
- 猜测
C^(0)(或P^(0))。 - 构建 P:使用
C^(k)计算密度矩阵P^(k)。 - 构建 F:使用
P^(k)构建 Fock 矩阵F^(k)(如左侧推导所示)。 - 构建 F’:使用
S^(-1/2)(它在计算开始前就算好了)和F^(k)来构建变换后的 Fock 矩阵F'^(k) = S^(-1/2) F^(k) S^(-1/2)。 - 求解:
F'^(k) C'^(k+1) = C'^(k+1) ε^(k+1)。这是一个标准本征值问题,计算机可以高效求解,得到新的C'和ε。 - 反变换:通过
C^(k+1) = S^(-1/2) C'^(k+1)得到我们真正的系数矩阵C。 - 检查收敛:用
C^(k+1)计算新的P^(k+1),与P^(k)比较。如果不收敛,返回第 2 步。
在数学上解决了如何在非正交基组下求解 HF 方程的实际计算问题。
III:
两个主要部分:
- 上部:完成 Roothaan-Hall 方程的数学求解变换。
- 下部:引入一个全新且至关重要的概念——Hartree-Fock (HF) 的总能量。
方程的最终求解形式
“标准本征值问题”变换的总结和补充。
S^(-1/2) = U† (1/√λ ... 0; 0 ... 1/λ_m) U- 这是
S^(-1/2)矩阵的明确计算方法,即通过对重叠矩阵S进行“谱分解”:- 对角化
S得到其本征向量矩阵U和本征值对角矩阵Λ(对角元为λ_i)。 - 计算
Λ^(-1/2)(即把每个λ_i替换为1/√λ_i)。 - 重新组合
U† Λ^(-1/2) U得到S^(-1/2)。
- 对角化
- 这是
(blas, lapack)- 这是一个非常实际的课堂笔记。
BLAS(Basic Linear Algebra Subprograms) 和LAPACK(Linear Algebra Package) 是用于高性能科学计算(如矩阵对角化、求逆等)的黄金标准软件包。 - 教授在这里的意思是:“这个矩阵运算(
S^(-1/2))我们手算不了,但计算机上的量子化学软件会调用LAPACK库来高效地完成它。”
- 这是一个非常实际的课堂笔记。
F' C' = C' ε- 最终方程。重申上一张白板的结论:我们已经成功地将广义本征值问题
F C = S C ε转换为了标准本征值问题F' C' = C' ε。 - 这是计算机可以(通过
LAPACK)直接求解的。
- 最终方程。重申上一张白板的结论:我们已经成功地将广义本征值问题
F' = S^(-1/2) F S^(-1/2) = (F')†- 这一行在确认一个重要的数学性质:
F'矩阵也是厄米 (Hermitian) 的。 †(dagger) 符号代表“厄米共轭”(转置并取复共轭)。- 因为
F是厄米的 (F† = F),S也是厄米的,所以F'保证是厄米的。这确保了我们求解得到的轨道能量ε必定是实数,这在物理上是必需的。
- 这一行在确认一个重要的数学性质:
Hartree-Fock (HF) 总能量
这是本张白板的核心。在 SCF 迭代收敛后,我们得到了所有的轨道能量
ε_i。那么,分子的总能量 E_HF
是多少?
一个常见的陷阱:你可能会认为总能量就是所有占据轨道的能量之和(2 Σ ε_i,因为每个轨道
2 个电子)。白板明确指出:这是错误的!
E_HF ≠ 2 Σ[i=1 to N/2] ε_i(总能量 ≠ 轨道能量之和)- 为什么?
- 白板上的最后一行给出了答案。轨道能量
ε_i的定义是:ε_i = <φ_i | f̂ | φ_i> = ε_ii + Σ[j=1 to N/2] (2J_ij - K_ij) - 物理含义:
ε_i不仅仅是电子i的能量,它代表了将一个电子加入到轨道i中所需要的能量。这个能量包括了:ε_ii:电子i自己的动能和它与所有原子核的吸引能。Σ[j] (2J_ij - K_ij):电子i与所有其他j轨道电子的库仑排斥和交换作用。
- 双重计算问题:
ε_i包含了i与j的排斥能。ε_j包含了j与i的排斥能。- 如果你简单地将它们相加
(ε_i + ε_j),你就把i和j之间的排斥能计算了两遍。
- 因此,
2 Σ ε_i会双倍计算所有的电子-电子排斥能,导致结果错误。
正确的 HF 总能量公式 白板给出了两个等价的正确公式:
E_HF = 2 Σ[i] ε_ii + Σ[i,j] (2J_ij - K_ij)(白板第一行)2 Σ[i] ε_ii:所有电子的动能 + 电子-原子核吸引能(ε_ii在这里是核心哈密顿积分h_ii)。Σ[i,j] (2J_ij - K_ij):所有电子对之间的排斥/交换能(只计算一次!)。
E_HF = Σ[i=1 to N/2] (ε_ii + ε_i)(白板中间行)- 这是一个更巧妙、更简洁的公式。
- 它将总能量表示为:对所有占据轨道
i求和,每一项是(核心哈密顿积分ε_ii+ 轨道能量ε_i)。 - 这个公式通过只加一次
ε_ii(单电子项)和一次ε_i(包含单电子项和双电子项),巧妙地修正了双重计算问题,最终结果与公式 1 完全等价。
总结
从“如何求解”到“如何获取最终能量”的过渡。它展示了求解
F C = S C ε 的实用计算方法,并着重强调了 HF 总能量
E_HF 和轨道能量 ε_i 之间的关键区别。
IV
它用一个清晰的流程图 (Flowchart),前面包含的所有复杂的数学公式和概念,总结成了一个完整的计算算法。
这就是 Hartree-Fock 自洽场 (Self-Consistent Field, SCF) 迭代循环的标准计算流程。
💡 SCF 流程图详解
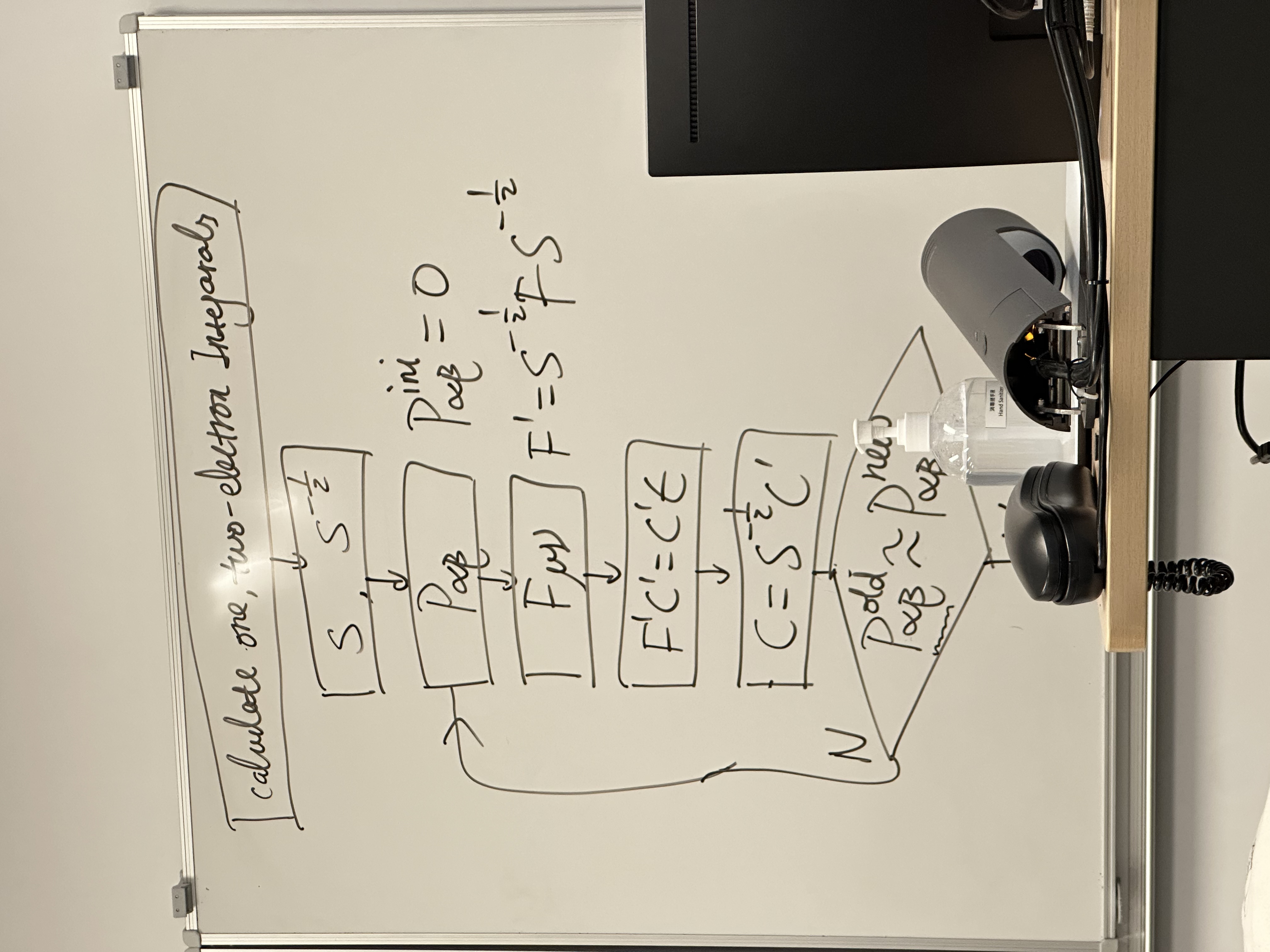
这个流程图展示了量子化学程序是如何一步步“猜”出正确答案的。
1. 准备工作 (循环开始前)
Calculate one, two-electron Integrals- 这是计算的“第 0 步”,在循环开始前只需要做一次。
- 程序会计算所有需要的“积木块”:
- 单电子积分:重叠矩阵
S和核心哈密顿矩阵H_core。 - 双电子积分:所有
(μν|αβ)形式的积分。这些积分数量极其庞大,是 HF 计算中最耗时的一步。
- 单电子积分:重叠矩阵
[ S -> S^(-1/2) ]- 利用第一步算出的
S矩阵,计算出用于正交化的S^(-1/2)矩阵。这也只需要做一次。
- 利用第一步算出的
2. SCF 迭代循环 (The Loop)
[ P_αβ ](起始点)- 第 1
步:猜测。循环开始,我们必须提供一个初始的密度矩阵
P。 - 白板上的
P_αβ^ini = 0是一个最简单的“零猜测”,实际程序通常会用更高级的猜测方法(如H_core猜测)。
- 第 1
步:猜测。循环开始,我们必须提供一个初始的密度矩阵
[ F_μν ]- 第 2 步:构建 Fock 矩阵。
- 使用当前的密度矩阵
P_αβ,根据我们在第二张白板上的公式F = H_core + G(P)来构建当前的 Fock 矩阵F。
[ F' C' = C' ε ]- 第 3 步:求解 Roothaan-Hall 方程。
- 正如第三张白板所示,我们不直接解
F C = S C ε。 - 我们先进行变换:
F' = S^(-1/2) F S^(-1/2)。 - 然后求解这个“标准本征值问题”,得到新的轨道能量
ε和变换后的系数C'。
[ C = S^(-1/2) C' ]- 第 4 步:反变换。
- 用
S^(-1/2)矩阵将C'转换回我们真正需要的、在原子轨道基组下的系数矩阵C。
[ P_αβ^old ~ P_αβ^new ](决策点)- 第 5 步:检查自洽性 (Convergence Check)。
- 使用第 4 步得到的新
C,计算出一个新的密度矩阵P^new。 - 比较这个
P^new和我们在第 2 步中使用的P^old。 N(No): 如果P^new和P^old差别很大(未收敛),则自洽尚未达成。- 循环:将
P^new作为下一次迭代的P,返回第 2 步([ F_μν ]),用这个新的P去构建新的F。
- 循环:将
Y(Yes,未画出): 如果P^new和P^old几乎完全相同(差值小于某个阈值,例如 10⁻⁸),则说明“自洽”达成!- 循环结束。
总结
以上内容从“为什么”(HF 近似)到“是什么”(HF 方程和矩阵)再到“怎么做”(SCF 流程图)。
SCF 流程是所有基于 Hartree-Fock 方法(以及更高级的后 HF 方法)的计算化学软件的核心算法。